
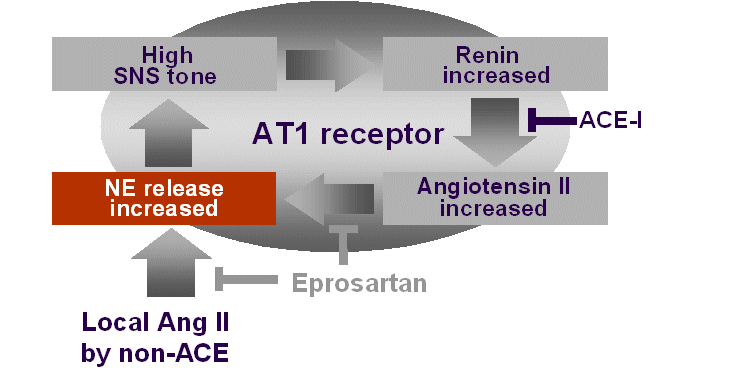
A hallmark of cardiovascular diseases is an adverse remodeling of the extracellular matrix involving an increased collagen deposition. The fibrosis results not only in an impaired pump function due to an increased stiffness of the myocardium but leads also to a reduced coronary perfusion. In the vasculature, a progressive remodeling is observed which appears to start in the arterioles leading to an increased diastolic and systolic hypertension. Particularly in the elderly, the remodeling reaches also the conduit arteries which store blood in the systole. Due to the reduced capacitive function of large arteries and the aorta, the diastolic blood pressure becomes normal while the systolic blood pressure is high, i.e. isolated systolic hypertension. This endstage of hypertension involves adverse effects of both catecholamines and angiotensin II (Ang II).
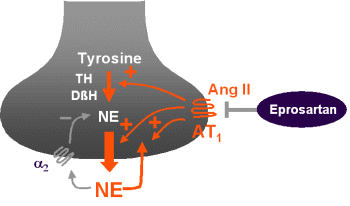
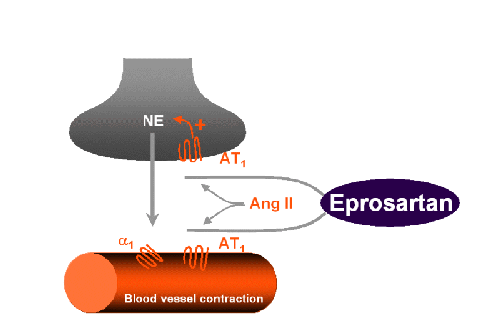
Ang II stimulates release of norepinephrine
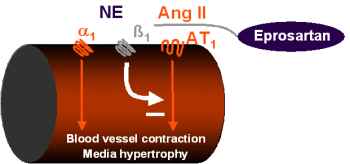
Therapeutic consequences
Eprosartan has a high
potency for inhibiting norepinephrine release
The deleterious effects of increased Ang II levels and catecholamines have traditionally been considered as two different therapeutic targets. Increasing evidence indicates, however, that drugs are available which affect both, the RAS and catecholamines. A lead compound of this promising class of drugs is the AT1 receptor blocker eprosartan (10). Eprosartan differs from currently used AT1 receptor blockers in various aspects. In contrast to the other AT1 receptor blockers, eprosartan has a distinct chemical structure (7). The results of preclinical studies to date indicate that the inhibition of sympathetic outflow by AT 1 receptor blockers is a class effect, with eprosartan being more potent than other agents in this class. This increased potency may be due to differences in the chemical structure and receptor binding characteristics of eprosartan when compared with other AT1 receptor blockers.
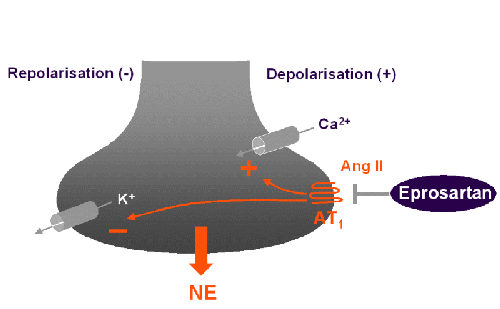
Ohlstein et al. (7) showed that the pressor responses to spinal cord stimulation were inhibited by the peptide Ang II receptor antagonist saralasin. This confirms the existence of prejunctional Ang II receptors at the vascular neuroeffector junction that facilitate release of norepinephrine. Furthermore, at a dose which completely blocked Ang II receptors (0.3 mg/kg, i.v.), the non-peptide AT1 receptor blocker eprosartan significantly inhibited the pressure response mediated by spinal cord stimulation. This high potency regarding presynaptic sympathetic inhibition in the pithed rat may be unique to eprosartan because other AT 1 receptor blockers such as losartan, valsartan and irbesartan failed to inhibit the pressure responses at the same dose (7). Recently, Balt et al. (2) determined the inhibitory potency of candesartan, valsartan, eprosartan and embusartan in blocking presynaptically and postsynaptically located Ang II receptors. The order of sympathoinhibitory potencies was eprosartan> valsartan = candesartan = embusartan (where > signifies P < 0.05). The order of potency of the agents tested for sympathoinhibition clearly differed from that for inhibition of the direct pressor effects of Ang II. It was concluded that considerable differences in affinity of the various AT 1 receptor blockers exist for pre- and postsynaptic AT 1 receptors.
Inhibition of raised sympathetic activity occurs at blood
pressure lowering doses
An inhibitory effect of eprosartan on sympathetic overactivity was also demonstrated in a model of hyperkinetic hypertension arising from a hypercaloric diet intake (9). The diet contained 24% fat and 32% sucrose which is expected to increase norepinephrine turnover in the heart (11). The radio telemetric data showed that during hypercaloric feeding, heart rate and blood pressure were significantly increased in conscious spontaneously hypertensive rats (SHR). Eprosartan not only prevented the blood pressure rise due to the hypercaloric diet intake but also consistently reduced the high blood pressure that is characteristic of SHR. The increased heart rate was also significantly reduced. Although an increased SNS activity is expected to raise the amount of circulating Ang II, the extent to which Ang II potentiated sympathetic nervous system activity during the hypercaloric diet intake has not yet been determined. It should be pointed out that the reduction in heart rate was observed at a dose of eprosartan that consistently reduced blood pressure. This indicates that the dose of eprosartan required to lower blood pressure also has an inhibitory effect on a raised heart rate. This is in contrast to observations in the pithed rat where the dose required for inhibition of sympathetic neurotransmission was generally higher than that needed for inhibition of an Ang II induced blood pressure response.
2. Balt JC, Mathy MJ, Pfaffendorf M, van Zwieten PA. Inhibition of facilitation of sympathetic neurotransmission and angiotensin II-induced pressor effects in the pithed rat: comparison between valsartan, candesartan, eprosartan and embusartan. J Hypertens 2001;19:2241-50.
3. Brasch H, Sieroslawski L, Dominiak P. Angiotensin II increases norepinephrine release from atria by acting on angiotensin subtype 1 receptors. Hypertension 1993;22:699-704.
4. Ihara M, Urata H, Kinoshita A, et al. Increased chymase-dependent angiotensin II formation in human atherosclerotic aorta. Hypertension 1999;33:1399-1405.
5. Lai KB, Sanderson JE. Effects of noradrenaline stimulation on collagen production and transforming growth factor-B1 gene expression in myocardial fibroblasts. J Amer Coll Cardiol 2002;39(Suppl. B):457B
6. Meloche S, Pelletier S, Servant MJ. Functional cross-talk between the cyclic AMP and Jak/STAT signaling pathways in vascular smooth muscle cells. Mol Cell Biochem 2000;212:99-109.
7. Ohlstein EH, Brooks DP, Feuerstein GZ, Ruffolo RR, Jr. Inhibition of sympathetic outflow by the angiotensin II receptor antagonist, eprosartan, but not by losartan, valsartan or irbesartan: relationship to differences in prejunctional angiotensin II receptor blockade. Pharmacology 1997;55:244-251.
8. Richards EM, Raizada MK, Gelband CH, Sumners C. Angiotensin II type 1 receptor-modulated signaling pathways in neurons. Mol Neurobiol 1999;19:25-41.
9. Rupp H, Benkel M, Maisch B. Control of cardiomyocyte gene expression as drug target. Mol Cell Biochem 2000;212:135-142.
11. Rupp H, Maisch B. Radiotelemetric characterization of overweight-associated rises in blood pressure and heart rate. Am J Physiol 1999;277:H1540-5.
12. van Zwieten PA, de Jonge A. Interaction between the adrenergic and renin-angiotensin-aldosterone-systems. Postgrad Med J 1986;62 Suppl 1:23-7.