Excess
Catecholamine Syndrome and metabolic syndrome
Hypertension is often associated with
metabolic disorders involving particularly insulin resistance. This
decreased insulin sensitivity leads to a compensatory hyperinsulinemia
that is commonly accompanied with hypertriglyceridemia and a raised
sodium and uric acid re-absorption of the kidney. In the last years,
these symptoms have been referred to as " metabolic syndrome" or
"syndrome X" (1). Characteristics of the metabolic syndrome are
hypertension, insulin resistance, hyperinsulinemia,
hypertriglyceridemia, hyperuricemia and reduced HDL-cholesterol.
The majority of persons with symptoms of the
metabolic syndrome are overweight. The PROCAM study has shown (2) that
in parallel to
the increase in body mass index those parameters are increased that
have been
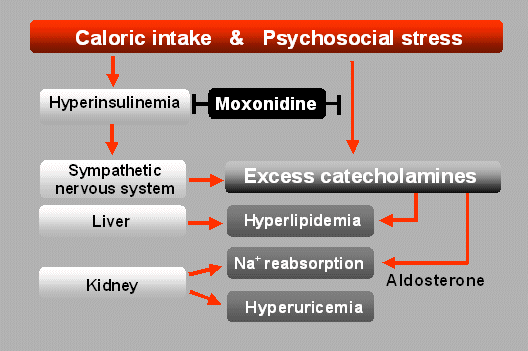
summarized under the term metabolic syndrome. A prolonged hypercaloric diet intake is
expected to
induce insulin resistance and hyperinsulinemia. An increased
sympathetic tone
can amplify insulin resistance involving several mechanisms, i.e. acute
and
chronic reactions. An acutely raised sympathetic activity increases the
glucose
output of the liver while the insulin secretion of the beta-cells of
the
pancreas becomes relatively diminished. If a teleological viewpoint
would
be justified then one could argue that this mechanism garantuees
the
supply of glucose for the insulin-independent glucose use of the brain
that
is mobilized by epinephrine when food shortage occurs. At the same
time, the
lipolysis induced increase in plasma fatty acids causes a further
reduction of peripheral glucose utilization ("Randle cycle").
Chronic modifications
in the morphology of skeletal muscle contribute to a further reduction
in insulin sensitivity. The arteriolar density of skeletal muscle
becomes reduced ("rarefaction") leading to increased diffusion
distances for oxygen and probably also glucose (3) after a chronic
beta2-adrenergic stimulation. Furthermore, the proportion of fast
fibers with a lower glucose oxidation becomes increased at the expense
of slow muscle fibers (4).
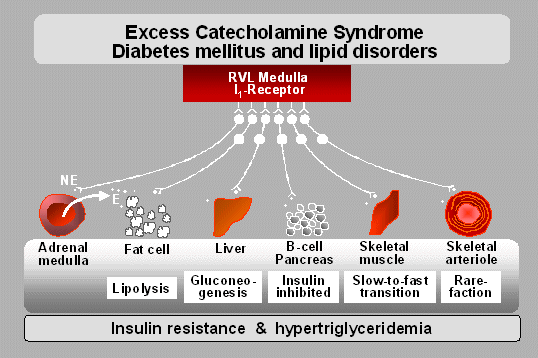
Although the hyperinsulinemia can
initially prevent hyperglycemia, this state can frequently not be
maintained and diabetes mellitus type II ensues. B-cells of the
pancreas are limited in their production
of insulin. Oral antidiabetic drugs lead to an increased insulin
release for preventing hyperglycemia. A consequence is that organs that
do not become
insulin-resistant are exposed to higher insulin levels.
The insulin resistance
of the body arises predominantly from a reduced insulin sensitivity of
skeletal muscle, i.e. it represents a selective insulin resistance.
Organs that are not insulin resistant are influenced by the higher
insulin levels. In the liver, an increased triglyceride and LDL
synthesis ensues and in the kidney the re-absorption of sodium and uric
acid is increased. The hyperinsulinemia associated reactions amplify,
therefore, direct effects of a raised sympathetic nervous system
activity.
Since the centrally
acting antihypertensive drug moxonidine
( Physiotens, Moxon,
Cynt, Moxol and Normatens)
was shown to increase insulin
sensitivity
of overweight insulin-resistant hypertensive patients (5) it is
expected
to reduce also other negative effects arising from hyperinsulinemia.
1. Reaven GM: Banting
lecture 1988. Role of insulin resistance in human disease. Diabetes
1988;37:1595-1607
2. Assmann G, Schulte H:
Results and conclusions of the Prospective Cardiovascular Münster
(Procam)
Study, in Assmann G (ed): Lipid Metabolism and Coronary Heart Disease.
München, MMV-Verlag, 1993, pp 19-67
3. Suzuki J, Gao M, Xie Z,
Koyama T: Effects of the beta 2-adrenergic agonist clenbuterol on
capillary geometry in cardiac and skeletal muscles in young and
middle-aged rats. Acta Physiol Scand 1997;161:317-326
4. Zeman RJ, Ludemann R,
Easton TG, Etlinger JD: Slow to fast alterations in skeletal muscle
fibers caused by clenbuterol, a beta 2-receptor agonist. Am J Physiol
1988;254:E726-32
5.
Haenni A, Lithell H: Moxonidine improves insulin sensitivity in
insulin-resistant hypertensives. J Hypertens 1999;17 Suppl 3:S29-35